A new cobalt-photoredox method from the Teskey group turns the internal carbon of an olefin into a nucleophile, delivering 2-arylpropanoic acids in one step from styrenes and atmospheric CO2.
Metal hydride hydrogen-atom-transfer (MHAT) catalysis is one of the most powerful tools in radical chemistry. A high-valent cobalt, manganese, or iron hydride delivers a hydrogen atom to an olefin with Markovnikov regiochemistry, generating a stabilized carbon-centered radical at the internal position. From there, the radical can be trapped by a π-system, coupled with a persistent radical, or oxidized to an alkyl-metal(IV) species that behaves as a carbocation equivalent and reacts with nucleophiles.
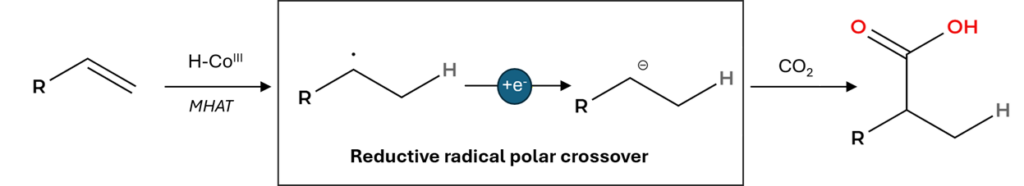
Every one of these pathways shares the same underlying logic: the internal carbon is electrophilic. The polarity matches what an undergraduate would predict from a classical Markovnikov addition.
In a recent paper in Chemical Science, Samikshan Jana, Daniel Kusza, Nikita Vystavkin, Danijela Lunic, and Christopher Teskey at TU Braunschweig report something genuinely different. Their work, « Inverting polarity in a cobalt MHAT reaction via reductive catalytic turnover, » is the first catalytic method that flips this polarity. The internal carbon becomes nucleophilic and reacts with an electrophile.
How the polarity gets inverted
The trick is to swap the oxidative turnover of the classical MHAT cycle for a reductive one. Instead of regenerating the active Co(III)-H species through oxidation of Co(II), the Teskey group uses photoredox catalysis to reduce Co(II) to Co(I), which is then protonated to give Co(III)-H. After hydrogen atom transfer to the styrene, the resulting benzylic radical is reduced a second time by the photocatalyst to give a benzylic carbanion. That carbanion is the nucleophilic intermediate that attacks CO2.
The result is an overall Markovnikov hydrocarboxylation: a styrene plus a balloon of CO2 delivers a 2-arylpropanoic acid, with the carboxylic acid installed at the internal carbon.
The optimized conditions are notably mild and use entirely commercial materials:
- 3.0 mol% (S,S)-Co(salen) tBu,tBu
- 1.0 mol% 4CzIPN photocatalyst
- 2.0 equivalents Hantzsch ester as the terminal reductant
- CO2 balloon (1 atm)
- DMF, 0.05 M, 450 nm irradiation, room temperature, 18 hours
No stoichiometric metallic reductants, no high-pressure CO2, no bespoke ligand synthesis.
A platform for 2-arylpropanoic acids and quaternary centres
The product class is significant. The 2-arylpropanoic acid (or α-aryl carboxylic acid) scaffold sits at the core of an entire family of nonsteroidal anti-inflammatory drugs, the profens: ibuprofen, ketoprofen, naproxen, flurbiprofen, fenoprofen, and others. The Teskey team showed the method can synthesize flurbiprofen directly from the corresponding styrene on 1 mmol scale in 65% yield.
The substrate scope spans more than 30 examples and tolerates a broad range of functional groups: aryl nitriles, sulfones, ketones, esters, aryl halides (including ortho-bromo), heteroarenes such as thiophene, and even unprotected ortho-anilines, which cyclize in situ to give substituted oxindoles.
Two features of the scope stand out for synthetic chemists.
First, the method constructs fully substituted quaternary α-aryl carbon centres from 1,1-diaryl styrenes in 75 to 90% yields. This is a motif that the closest existing nickel-catalyzed hydrocarboxylation (König and coworkers) cannot access, and it remains difficult to make by most other routes.
Second, the carbonyl-transposition strategy is elegant. Taking a diaryl ketone-containing drug such as fenofibric acid or ketoprofen, performing a Wittig olefination, and then applying these hydrocarboxylation conditions formally transposes the carbonyl by one atom while installing a new quaternary stereocentre. The acid handle was also appended to estrone, tigogenin, tryptamine, and leelamine, demonstrating real late-stage diversification.
Distinct from nickel, distinct from classical MHAT
The Teskey cobalt method is mechanistically orthogonal to the established nickel-catalyzed hydrocarboxylation. The nickel system proceeds through concerted hydrometallation and C-C bond formation from an organonickel intermediate. The cobalt system proceeds through a discrete benzylic carbanion, supported by three lines of evidence: the product is racemic despite the use of an enantiopure cobalt catalyst, a cyclopropyl radical clock substrate opens cleanly, and MHAT is shown to be irreversible by recovery of non-deuterated starting material.
The practical consequences are significant. The cobalt method tolerates aryl chlorides and bromides (which are dehalogenated by typical 4CzIPN systems), constructs quaternary centres that nickel cannot, and works on β-substituted styrenes.
The selectivity is also unusual in the opposite direction. When a substrate contains both a styrene and a tethered 1,1-disubstituted aliphatic olefin (the classical preferred MHAT acceptor), only the styrene reacts. Acrylates are not hydrocarboxylated either. The photoinduced reductive MHAT pathway therefore ignores olefins that classical oxidative MHAT would happily consume, which makes it useful as a chemoselective tool in densely functionalized molecules.
Running it in the EvoluChem PhotoRedOx Box Duo
The reactions were carried out in the EvoluChem PhotoRedOx Box Duo with 450 nm 18W EvoluChem PF LEDs. The Duo’s two-LED design and 16-vial capacity is well-matched to the kind of work this paper required: a 30+ substrate scope, condition optimization across multiple variables (Co loading, photocatalyst loading, reductant equivalents, proton source), and detailed mechanistic studies including deuterium-labeling experiments with three different Hantzsch ester isotopologues. Running these in parallel with reproducible photon flux at 450 nm is exactly what the PhotoRedOx platform is built for.
For groups looking to enter this kind of chemistry, the entry-level EvoluChem PhotoRedOx Box uses the same patented chamber design and the same EvoluChem LED ecosystem, scaled to a single LED and 8 vials. It is a straightforward, stir-plate-compatible setup for a first pass at photoredox conditions.
A new transformation for the toolbox
What makes this paper worth a careful read is that it does not just add a new reaction. It opens a new mode of reactivity for MHAT chemistry. Reversing the polarity of the olefin under MHAT conditions had been an unsolved problem for the field; the only previous example required a stoichiometric chromium salt. By coupling a photoredox-driven reductive cobalt cycle to radical-polar-crossover, the Teskey group has built a general, catalytic platform for installing electrophiles at the internal carbon of a styrene.
For medicinal chemists, the immediate payoff is direct access to 2-arylpropanoic acid pharmacophores, including quaternary-centre variants, with broad functional group tolerance and late-stage compatibility. For photochemists and methodology groups, the underlying logic of reductive MHAT plus RRPC is a new design space.
We are looking forward to seeing where this goes next.
Explore the EvoluChem PhotoRedOx Box Duo
Double the LED capacity. 16 vials in parallel. Same patented chamber design.
Reference: S. Jana, D. A. Kusza, N. Vystavkin, D. Lunic, C. J. Teskey, Chem. Sci. 2026, 17, 1098-1104. DOI: 10.1039/d5sc06119a
Equipment used: EvoluChem PhotoRedOx Box Duo with 450 nm 18W EvoluChem PF LEDs.
