Researchers at the Moffitt Cancer Center and the University of South Florida report a metal-free photoredox strategy for C–H amination of unactivated substrates using diazirines as the nitrogen source – including the first synthesis of acyldiaziridines from simple aldehydes, run throughout in the HepatoChem PhotoRedOx Box.
Nitrogen-containing compounds are woven through every corner of chemistry. According to a 2024 analysis in the Journal of Medicinal Chemistry, the share of newly approved U.S. FDA small-molecule drugs containing at least one nitrogen heterocycle has climbed from 59% to 82% over the past decade. Yet the direct introduction of nitrogen onto unactivated carbon–hydrogen bonds, bypassing the need for pre-functionalized starting materials, remains a genuinely difficult problem. Intermolecular C–H amination of inert aliphatic substrates has historically required transition metals, large excesses of reagents, or harsh conditions that limit functional group compatibility.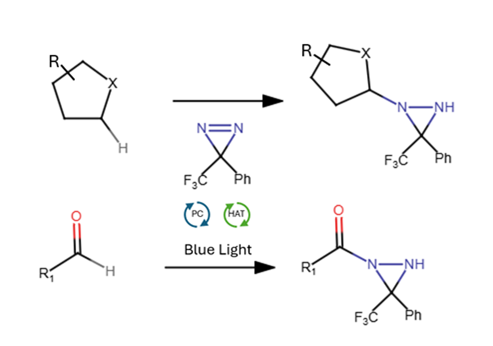
A new paper in Organic Letters from Daria V. Galaktionova, Vishala Maharaj, and Justin M. Lopchuk at the H. Lee Moffitt Cancer Center and Research Institute and the University of South Florida describes a practical solution. Their method uses a dual acridinium and DABCO-based hydrogen atom transfer (HAT) photocatalytic system to achieve C–H amination of unactivated substrates across four different substrate classes under mild conditions, without any transition metal catalyst. The diazirine serves as a radical acceptor rather than the carbene precursor it is more commonly known as, giving clean access to diaziridine products in up to 95% yield across 40 examples. Most strikingly, the method provides the first access to acyldiaziridines from aldehyde feedstocks, a previously unavailable class of compounds that function as versatile synthetic intermediates.
All reactions throughout the study were run in the HepatoChem PhotoRedOx Box equipped with a 456 nm blue LED.
The catalytic system: acridinium photocatalyst meets DABCO-HAT mediator
The key insight driving the method is the combination of two distinct catalytic functions. The acridinium photocatalyst, 3,6-di-tert-butyl-2,4,6-trimethyl-10-phenylacridinium tetrafluoroborate (t-Bu₂-Mes-Acr⁺BF₄⁻, used at 5 mol%), absorbs visible light and generates a powerful excited-state oxidant capable of initiating radical chemistry. The DABCO-based HAT catalyst (DABCO-cat3, used at 10 mol%) mediates hydrogen atom transfer from the C–H substrate, generating a carbon-centered radical.
That carbon radical then adds to the diazirine. Critically, the diazirine functions here as a radical acceptor via C–N bond formation, not as a carbene precursor, the role it plays in most photochemical applications. No cyclopropane fragmentation products were detected at any point during the study, and re-exposure of isolated diaziridine product to the reaction conditions caused no decomposition or further reaction, confirming that the N–H bond of the diaziridine is incompatible with the catalytic cycle. The reaction is carried out in acetonitrile under an argon atmosphere at 20–30 °C, with irradiation at 456 nm for 24–48 hours.
Screening of photocatalysts and HAT mediators revealed that 4CzIPN and DABCO were incompatible, while the acridinium/DABCO-cat3 combination was optimal. Degassing the catalyst mixture in acetonitrile before adding the substrate was critical: oxygen suppresses the reaction completely, and Ar-purging prior to substrate addition (rather than after) was important for reproducibility.
Four classes of C–H bonds, one general set of conditions
With the optimized conditions established, the Lopchuk group explored the scope across four substrate classes.
Unactivated cyclic and acyclic hydrocarbons are the most challenging class. Cycloalkanes of different ring sizes (cyclopentane through cyclooctane) gave diaziridines in good to excellent yields. Adamantane reacted with complete selectivity at the tertiary position (57% yield), while cis-decalin gave a mixture of tertiary and secondary diaziridines; trans-decalin gave only secondary products, consistent with steric control of the HAT step. The reaction was scaled to 4 mmol without any loss in yield, producing 1.12 g of cyclooctane diaziridine product. A tertiary bromide was tolerated as a substrate, as were ketones and secondary and tertiary alcohols – all giving diaziridines with good regioselectivity in the expected sense (less hindered tertiary C–H bonds preferred).
α-Heteroatom C–H bonds (adjacent to oxygen) are generally weaker than unactivated aliphatic C–H bonds and prove to be excellent substrates. THF, 2-Me-THF, 1,4-dioxane, diethyl ether, and more complex natural-product-derived ether substrates (reduced cyrene, isomannide, ambroxide) all reacted cleanly, with selectivity for the least substituted carbon adjacent to oxygen in the case of 2-substituted THF derivatives.
Benzylic C–H bonds react predictably, with indane giving 89% yield at 10 equivalents of substrate. Carbonyls β to the benzylic position also participated selectively.
Acyl C–H bonds from aldehydes represent the genuinely new advance. Benzaldehyde furnished acyldiaziridine 26 in 65% yield, scalable to 2 mmol. The reaction tolerates both electron-rich (p-OMe) and electron-poor (p-Br, p-Cl, p-CO₂Me) aromatics, ortho-halide substitution (though ortho-bromo shows some diminution), and alkyl and α,β-unsaturated aldehydes. Crotonaldehyde and simple aliphatic aldehydes gave yields up to 99%. The product is a previously inaccessible compound class: the acyldiaziridine, in which a nitrogen-nitrogen bridge sits directly on a carbonyl.
Standard conditions: t-Bu₂-Mes-Acr⁺BF₄⁻ (5 mol%), DABCO-cat3 (10 mol%), CH₃CN, Ar atmosphere, 456 nm blue LED, 20–30 °C, 24–48 h
Scale: Demonstrated at 4 mmol (1.12 g product from cyclooctane)
Photoreactor: HepatoChem PhotoRedOx Box
Acyldiaziridines: a new class of versatile intermediates
The most synthetically important finding in the paper may be what you can do with acyldiaziridines once you have them. The Lopchuk group demonstrated a broad set of transformations from acyldiaziridine 26 (from benzaldehyde), establishing these molecules as what the authors call “masked amides” obtainable from simple aldehyde feedstocks in a single step.
Treatment with hydroiodic acid in acetonitrile at room temperature converts acyldiaziridine 26 to amide 42 in quantitative yield. Hydrazide salt 43 is obtained in 96% yield with p-toluenesulfonic acid in dichloroethane at 90 °C. From the hydrazide, two mixed diacylhydrazides were synthesized using an acyl chloride and a carboxylic acid coupling partner – diacylhydrazide substructures that appear in a class of pesticide compounds. Rapid SNAr reactivity of acyldiaziridine 26 under acidic conditions gave acyl/aryl mixed hydrazide 46 in 80% yield. The acyldiaziridine also undergoes ring-opening to give oxadiazole 47 in one step from acyl chloride – oxadiazoles are found in approved drugs and have been validated as bioisosteric replacements for esters and amides. Acylhydrazone 48 was formed cleanly by heating in dioxane without purification.
Perhaps most practically significant is the discovery that acyldiaziridines serve as coupling partners in copper-catalyzed cross-coupling with aryl and vinyl halides, adapted from Klapars and Buchwald conditions. Both bromides and iodides reacted well; chlorides were inert. The resulting amides include an HDAC8 inhibitor precursor (with ester tolerance confirmed) and a celecoxib analogue made by late-stage functionalization. Alkyl and vinyl acyldiaziridines were also competent coupling partners. The authors demonstrate the synthesis of an SK2 kinase inhibitor precursor in 43% overall yield from 4-chlorobenzaldehyde in only two steps.
The HepatoChem PhotoRedOx Box as the reaction platform
Every reaction in this study – from the initial optimization screen through the 40-example substrate scope and all downstream diversification reactions – was run in the HepatoChem PhotoRedOx Box equipped with a 456 nm blue LED at 100% intensity. The vial format of the PhotoRedOx Box (8 mL reaction vials at 0.1–0.2 M in 2 mL acetonitrile for screening, scaled to 4 mL–20 mL vials for larger runs) is directly suited to this class of photocatalytic C–H functionalization reactions, where parallel screening of catalyst/HAT mediator combinations and consistent irradiation geometry are both important for reliable optimization. The EvoluChem 450PF LED provides the equivalent visible-wavelength irradiation for groups wishing to reproduce this chemistry with EvoluChem light sources.
If you are looking to explore acridinium-catalyzed photoredox HAT chemistry or to adapt the diaziridination conditions described here, the PhotoRedOx Box provides the standardized platform used in this work.
Explore the HepatoChem PhotoRedOx Box
The photoreactor used in this study. Parallel vial formats, interchangeable EvoluChem PF LEDs, compatible with both screening and gram-scale preparative runs.
Reference: D. V. Galaktionova, V. Maharaj, J. M. Lopchuk, Org. Lett. 2026, 28, 7315–7320. DOI: 10.1021/acs.orglett.6c01782
Equipment used: HepatoChem PhotoRedOx Box with 456 nm blue LED.
